有机化学补天笔记
一些广为人知的基础知识就略过了
Ch1 有机化合物分子结构基础
基本都是高中化学选修内容
共价键的极性
不同杂化轨道碳原子的电负性(酸性)大小顺序为:
诱导效应
吸电子诱导效应
多为带有双键的集团和卤素(拟卤素)原子
给电子诱导效应
共振式
共振结构的书写
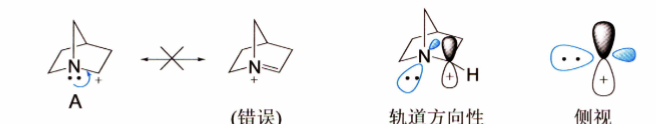
- 只有电子可以离域,不能改变分子的骨架
- 注意满足轨道方向性的要求,例如:
Ch2 脂肪烃和脂环烃
脂肪烃的命名
- 碳链最长原则
- 重键最多原则
- 双键最多原则
- 重键位次组最低原则
还有好多乱七八糟的,不想打字了
脂肪烃的结构
烯烃构型的命名
- 顺反命名法:相同的基团在同侧,称为顺式(cis-);否则称为反式(trans-)
- 次序规则
- 原子序数大的优先于原子序数小的
- 原子质量高的优先于原子质量低的
- 顺优先于反,Z优先于E
- R优先于S
和之后的 R/S 命名法一样
- Z/E 命名法:较优的基团在同侧,称为Z(Zusammen,同)构型;否则称为E(Entgegen,反)构型
环烷烃的构象
环己烷的构象
- 直立键(axial bond / a 键)
- 平伏键(equatorial bond / e 键)
环丙烷
在环丙烷中,C-C-C 键角不是 60 度,否则张力会很大,接近 105 度
Ch3 对映异构
旋光性 分子的手性
分子的手性与对称性
如果分子存在对称面或对称中心,则一定无手性,但是存在对称轴,不一定
Fischer 投影式
横键朝前,竖键朝后
构型的标记
- R/S 标记法:使最小基团远离观察者,若剩下三个基团从大到小为顺时针,称为 R 构型(Rectus,右);否则称为 S 构型(Sinister,左)
- D/L 标记法
Ch4 碳碳重键的加成反应
烯烃和炔烃的亲电加成反应
- 碳碳重键的电子进攻亲电试剂,形成碳正离子中间体(决速步)
- 亲核试剂捕获碳正离子,形成加成产物
马可尼可夫规则 Markovnikov Rule
氢总是加到氢多的碳上,本质上是因为氢少的一端容易形成碳正离子中间体(因此,双键碳上的烷基越多,反应速率越快)
碳正离子
碳正离子是缺电子结构,会被给电子基团 EDG 稳定,被吸电子基团 EWG 不稳定
与卤素的加成
先形成卤𬭩离子中间体(也可以认为是卤原子稳定了相邻的碳正离子),卤负离子再从后方进攻——反式加成
共轭双烯的亲电加成
- 低温:1,2-加成,动力学控制产物
- 高温:1,4-加成,热力学控制产物
羟汞化-还原反应
汞正离子机理,生成马式产物,反式加成
硼氢化-氧化反应
四元环过渡态机理(负氢加成),生成反马式产物,顺式加成
常用硼氢化试剂:
Diels-Alder 反应
协同反应机理,顺式加成,正对负,负对正,双烯体必须处于顺式构象
详细的 D-A 反应将在 Ch17 讨论
烯烃与炔烃与氢的加成反应
烯烃的催化氢化
催化剂:Pt,Pd,Ni,Raney 镍,Pd/C
顺式加成
炔烃的催化氢化
催化剂:Lindlar 催化剂(中毒的Pd)
顺式加成到烯烃
炔烃的金属还原
试剂:Na 或 Li/液氨
反式还原为烯烃,负离子自由基机理
烯烃与炔烃与氧的亲核加成反应
炔烃更容易发生亲核加成,区域选择性主要受到位阻的影响
烯烃与炔烃与氧的加成反应
双羟基化反应
- 稀冷高锰酸钾:经历五元环高锰酸酯中间体,顺式加成为邻二醇
- 四氧化锇:顺式加成为邻二醇
- 碘-醋酸银氧化
- Prevost 反应:无水,碘的四氯化碳溶液与等摩尔的醋酸银,水解后生成反式邻二醇
- Woodward-Prevost 反应:亲电加成机理,有水环境下,生成顺式邻二醇
环氧化反应
常用试剂:m-CPBA
Ch5 自由基反应
自由基的产生
键的均裂:光解,热解,自由基引发剂(过氧苯甲酰 BPO,偶氮二异丁腈 AIBN)
自由基的结构及稳定性
甲基自由基比甲基碳正离子稳定
给电子基团和吸电子基团都能很好的稳定自由基
不饱和烃的 -H 卤代反应
烯丙位自由基机理
溴代试剂:NBS
卤代烃的脱卤反应
常用试剂:三丁基氢化锡
烯烃和炔烃与溴化氢的自由基加成
与亲电加成不同的是,引入了过氧化物,反应会走自由基机理,生成反马式产物
Ch6 芳香烃
苯环上的亲电取代反应
苯环不易发生加成,易发生亲电取代——芳香烃亲电取代反应()
络合物-苯𬭩离子机理
Friedel-Crafts 烷基化反应
卤代烃 + Lewis 酸,烷基正离子机理,易重排
Friedel-Crafts 酰基化反应
酰卤 + Lewis 酸,酰基正离子机理,由于酰基吸电子,一般不会二次取代
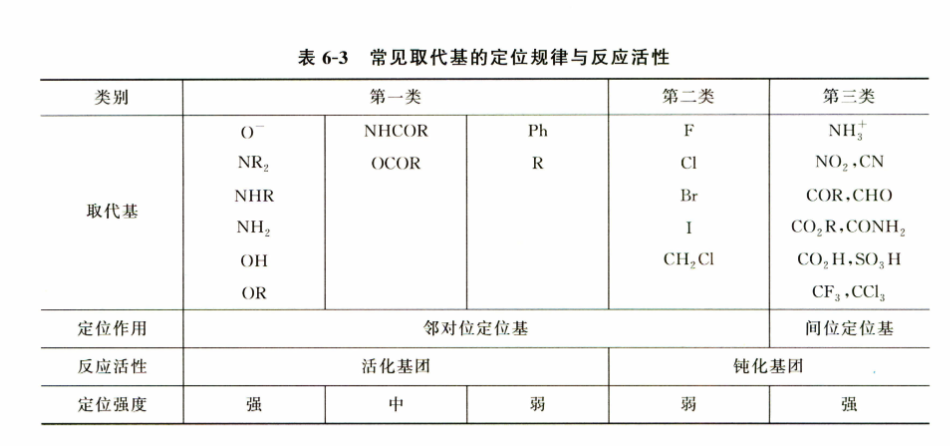
亲电取代反应的定位规律和反应活性
芳烃的氧化还原反应
还原反应
Birch 还原:Li-液氨-醇,苯被还原成环己-1,4-二烯
芳香性
Huckel 规则:闭环的,单双键交替的,共平面的, 的
Ch7 有机波谱分析基础
没讲,也希望别讲
Ch8 卤代烃
卤代烷烃的亲核取代反应
亲核取代反应
亲核取代反应
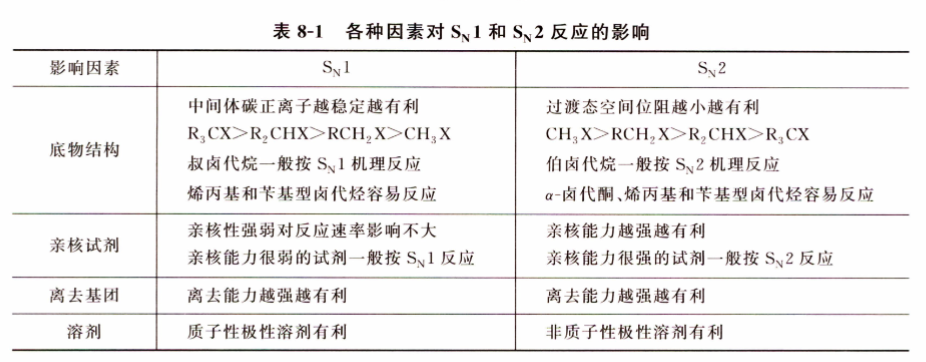
饱和碳原子上亲核取代反应的机理与立体化学
- 双分子亲核取代反应 :协同机理,发生构型反转,发生趋势与过渡态能量有关(主要是位阻,因此一级卤代烃活性最高)
- 单分子亲核取代反应 :碳正离子机理,得到构型保持和构型翻转两种产物(理论上1:1,实际上翻转略多),发生趋势与碳正离子稳定性有关(因此三级卤代烃活性最高),由于涉及碳正离子,因此会有重排产物
- 邻基参与 NGP:两次构型翻转,邻基需要在离去基的反式位置
影响亲核取代反应速率的因素
- 离去基的离去能力
- 烃基的结构
- 亲核试剂的亲核性:只有较强的亲核试剂才会走
- 溶剂
亲核性与两个因素有关:碱性(给电子能力);和可极化性
强碱强亲核:
强碱弱亲核:
弱碱强亲核:
卤代烷烃的消除反应
双分子消除反应 E2
碱进攻-H,经过过渡态直接生成E2消除产物
Zaitsev 规则:主要消除氢少的碳上的氢(不绝对,与碱的体积,产物的稳定性有关)
E2 必须是反式共平面才能消除(无视 Zaitsev 规则)
单分子消除反应 E1
弱碱的情况下会走 E1
- 生成碳正离子中间体(会有重排)
- 碱进攻-H,生成 E1 产物
单分子共轭碱消除反应 E1cb
弱离去,-H强酸
- 碱攫取-H,形成碳负离子中间体(共轭碱)
- 负离子推动离去基离去
消除反应与取代反应的竞争
| 因素 | E1 | E2 | ||
|---|---|---|---|---|
| -C 取代基越多 | + | - | + | + |
| 试剂碱性越强,体积越大 | + | - | ||
| 试剂碱性越弱,亲核性越强 | - | + |
卤代芳烃的亲核取代反应
加成-消除机理
当卤代芳烃的邻对位连有强吸电子基团,可以发生芳香亲核取代反应
苯炔机理
没有强吸电子基团,但是有强碱作为亲核试剂,走消除-加成机理(苯炔机理)
- 强碱攫取邻位氢形成苯基负离子
- 负离子推动卤离子离去,形成苯炔
- 强碱作为亲核试剂加成到苯炔上
卤代烃与金属反应
有机镁化合物的制备
Grignard 试剂 / 格氏试剂
无水溶剂(乙醚)
有机锂化合物的制备
无水溶剂中,双键构型保持,Li 取代 X